Strictly speaking immunophenotyping refers to the use of antibodies to different cellular markers to identify cell types through a hierarchical combination. It’s like antibody bar-coding a cell for positive and negative markers. Most often this involves using several surface and/or intracellular markers to identify subsets of white blood cells (i.e. Tregs) since they are largely morphologically indistinct spherical cells. When running blood cells it is common to eliminate RBCs as they are the vast majority of whole blood and make it impossible to “see” the peripheral blood mononuclear cells (PBMCs).
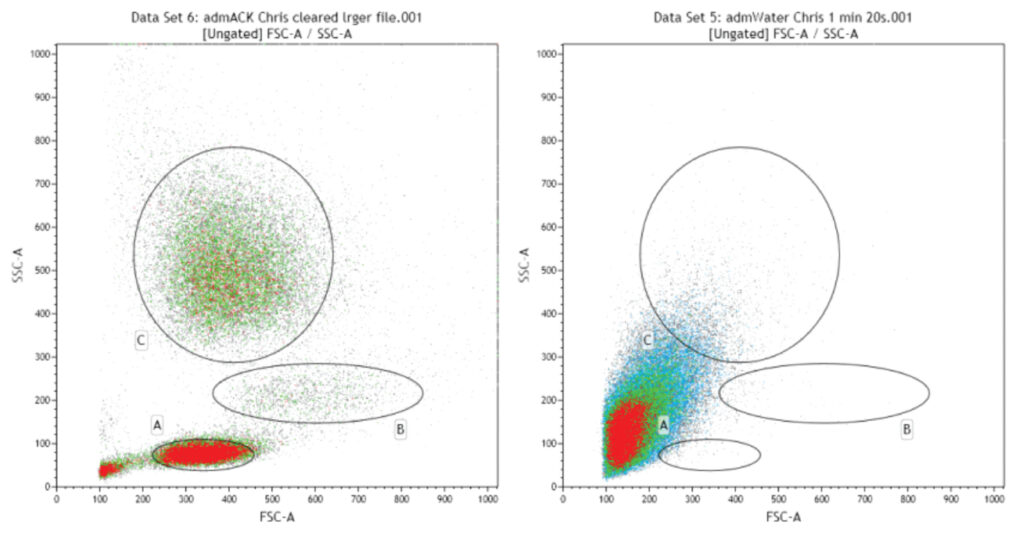
PBMCs can be roughly differentiated into 3 main populations on FSC/SSC (left panel: A- Lymphocytes, B- Monocytes and C- Granulocytes). If RBCs are not eliminated from the sample they overwhelm the PBMC minority and make analysis of these cells impossible (as shown by the identical sample pre-RBC lysis, right panel).
RBCs can be eliminated by several mechanisms including ACK (ammonium) lysis, water lysis (both lyse the RBCs preferentially via distinct mechanisms) and CD45 fluorescent thresholding (whereby CD45 labels all differentiated hematopoetic cells except RBCs and platelets, allowing for them to be “ignored” instead of lysed). A summary of these protocols and pros and cons of each approach can be found in Protocol 1:Facilities-Protocol1-PBMC-prep.
Subsequent multicolour staining will be highly dependent on the target cell population of interest (T-cell subset, progenitor cell, etc.). For many cell types there is an OMIP (online multicolour immunophenotyping panel) which have recommended staining parameters (antibody clones, concentrations, fluorophores). Often these OMIPs will discriminate between subsets beyond what you need (i.e. specific NK subsets and activation state when all you want is total NK cells). In that case you can be selective as to what subset markers you use or seek the advice of the core facility staff.
With all motile cells, capping can occur if the antibody crosslinks surface receptors. This leads to antigen shedding and cell death but this can be blocked via the inclusion of 0.01% Sodium Azide. Note that you cannot look at cell stimulation or energy dependent processes if you include azide.
In conjunction with the above recommendations, immunophenotyping then becomes multicolour flow cytometry specific to the experiment in question. As such there is no general protocol that can be provided, but general rules are as follows:
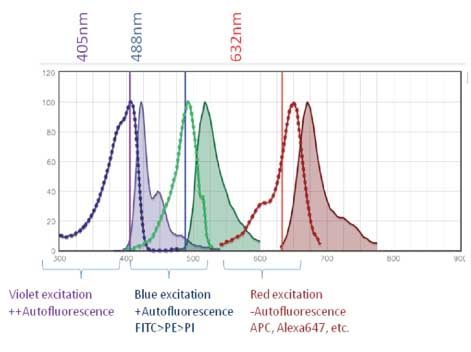
Short wavelengths (higher energy) are able to excite electrons on biological molecules within cells more easily than long wavelengths (lower energy). As a result, channels off of the 405nm laser and 488nm laser exhibit worse autofluorescence. This is worth keeping in mind when selecting a staining panel as this higher background can limit the resolution of dim/low abundance antigens.
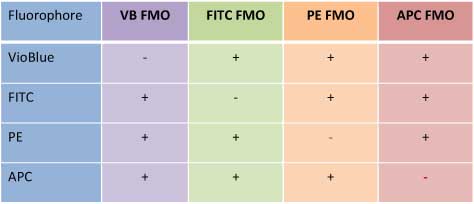
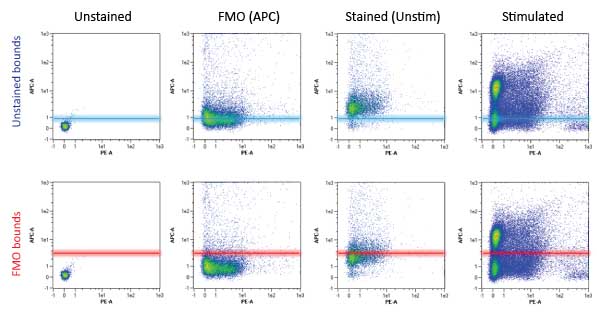
FMO Controls (Fluorescence Minus One) are done by excluding only the fluor of interest (i.e. a FITC FMO is stained for all markers except the FITC conjugated antibody). This allows for accurate setting of gating boundaries for the FITC channel. The FMO controls would be done for all fluors in the panel (see table to right) in an experiment using >3 colours. As you can see from the figure below, FMOs are crucial in setting these gating boundaries. Note that the “Stained (Unstimulated)” population using the unstained control boundaries (top row, 3rd panel) is falsely considered positive for the APC channel. With the APC FMO gating boundaries (bottom row, 3rd panel) this unstimulated population is correctly considered negative for the APC channel (in this case a marker of NK cell activation).