Staining intracellular antigens (phospho-proteins, transcription factors, etc) involves fixing the cells so that the intracellular contents do not float away and permeabilization of the cell membrane so that the antibodies/fluorochromes can enter the cell and label their targets of interest. The two most common ways of doing this are either a one-step fix/perm protocol (ethanol/methanol/commercial fix/perm reagents) or a two-step fix/perm protocol (most often PFA/Saponin) both of which are detailed in the respective protocols available on this site. The general pros and cons are:
Methanol/ethanol fixation (see Facilities-Protocol1-Onestep-Fix-perm) is very quick, fixes and permeabilizes at the same time and is very inexpensive. However, alcohols can lead to lipid extraction and removal of some protein and generally does not preserve intracellular structures well which is one of many reasons it is used less in microscopy.
PFA/Saponin fixation and permeabilization (see Facilities-Protocol2-Two-step-Fix-perm) is milder in this regard and tends to preserve epitopes and more “loosely” membrane-bound proteins. This process requires that saponin is in all wash steps as saponin pores are transient and you need to wash unbound antibody out of the permeabilized cell. In either case you will need an appropriate negative control as the permeabilized cell has more surfaces for non-specific binding and can passively trap unbound antibody inside. In most cases the appropriate control is an isotype control which is the same species, antibody type (i.e. mouse IgG1k), and has the same fluorophore in the same fluorophore/antibody ratio (i.e. Alexa488 cannot be used in place of FITC and if your antibody has 10 FITC moieties attached to it, your isotype control cannot have 20 FITC moieties attached to it).
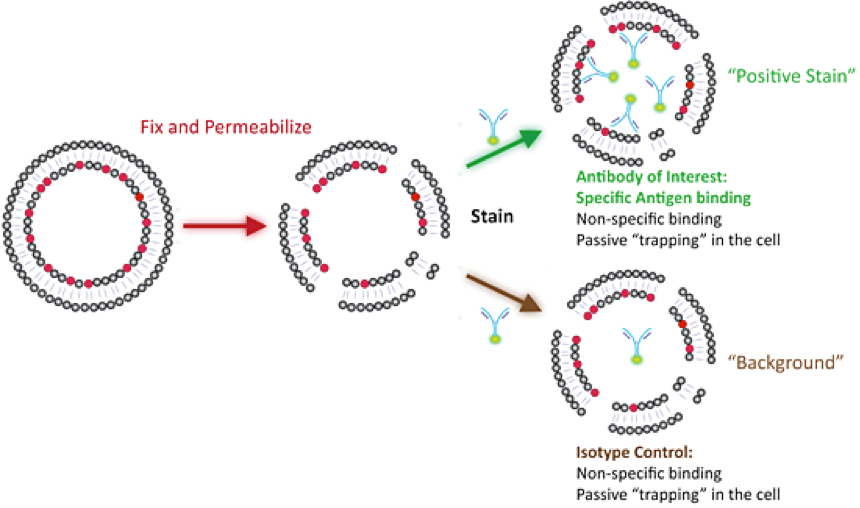 As is the case with any fixation step, both of these protocols will result in changes to epitopes and antigens in different ways. If it is unknown what fixation method works best for your antibody/antigen pair then it is best to do a small pilot experiment to determine which fixative results in the best positive staining and low background. For many markers there are compatibility resources for fixatives which can help save you time (see the fixation compatibility links).
As is the case with any fixation step, both of these protocols will result in changes to epitopes and antigens in different ways. If it is unknown what fixation method works best for your antibody/antigen pair then it is best to do a small pilot experiment to determine which fixative results in the best positive staining and low background. For many markers there are compatibility resources for fixatives which can help save you time (see the fixation compatibility links).
Other than the specifics of fixation and permeabilization the two main challenges of intracellular staining are 1) getting the antibody in to stain the antigen well and washing unbound background antibody out, and 2) sensitivity (increased background, autofluorescence, low signal due to epitope fixation). To this end many of the multicolour staining rules from the Immunophenotyping section are relevant. In particular: